Introduction aux GLMM avec données de proportion
Table des matières
Contexte
Le plan expérimental
Les données analysées dans ce tutoriel sont issues d’une expérimentation qui vise à évaluer les effets de deux traitements sur la survie ou la mort de cellules. Pour chaque traitement (traitement A ou traitement B), 4 boites de cultures sont utilisées (4 boites avec des cellules ayant reçu le traitement A et 4 boites avec des cellules ayant reçu le traitement B).
Pour chacune de ces boites, un certain nombre de photos sont prises (entre 6 et 24) sur une petite zone de la boite, et pour chaque photo, le nombre de cellules mortes et vivantes sont observées.
Le plan expérimental peut être résumé par ce schéma :

La question posée
La question qui est posée est « Est ce que les proportions (ou pourcentages) de cellules vivantes sont différentes selon le traitement reçu ? » Autrement formulé, « est ce qu’un des traitements est associé à une proportion plus importante de cellules vivantes ? »
D’un point de vu pratique, on cherche donc à faire une comparaison de deux pourcentages.
Caractéristiques des données et approche statistique
Données bornée
Il existe grossièrement trois types de GLM:
- les GLM avec lien log et distribution d’erreur de poisson lorsque les réponses observées sont de type comptage,
- les GLM avec lien logit et distribution d’erreur binomiale lorsque les réponses observées sont binaires (oui/non, mâle/femelle),
- les GLM avec lien logit et distribution d’erreur binomiale lorsque les réponses observées sont des proportions. C’est ce dernier modèle qui nous intéresse.
Données non indépendantes
Néanmoins, les données possèdent une autre caractéristique : elles ne sont pas indépendantes. En effet, le plan expérimental nous laisse penser que, pour un traitement donné, les pourcentages de cellules vivantes observées dans une même boite sont plus liés entre eux (corrélés) que les pourcentages provenant de deux boites différentes.
Pour comprendre ce concept, on peut imaginer, par exemple, qu’une des boites à un défaut de fabrication qui entraîne une sur-mortalité des cellules. Dans ce cas, toutes les observations réalisées sur cette boite sont impactées. Et donc les pourcentages de cellules vivantes observées
sur les images de cette boite sont plus semblables entre eux que ceux observées sur les images de deux boites différentes.
C’est cette seconde caractéristique, la non-indépendance, qui nous dirige finalement vers l’utilisation d’un GLMM (Generalized Linear Mixed Model ou modèle linéaire généralisé mixtes). Pour faire simple Un GLMM (Generalized Linear Mixed Model ou modèle linéaire généralisé mixtes) est un GLM avec une fonctionnalité supplémentaire qui lui permet de prendre en considération la non indépendance des données. Ici, on va donc utiliser un GLMM avec un lien logit et une distribution binomiale des erreurs.
Remarque très importante :
Utiliser un GLMM, et non pas un GLM, en cas de non indépendance des données ne relève pas du détail ! C’est CAPITAL ! Sans cette prise en compte de la corrélation entre certaines données, la déviance résiduelle du modèle employé pour analyser les données sera biaisée. Cela conduira à une estimation, elle aussi biaisée, de l’erreur standard des paramètres, et en bout de chaîne à une p value erronée.
A propose des GLMM
Pourquoi "mixte"
Un GLMM est dit “mixte”, car il comporte au moins un effet dit “fixe” (la variable dont on souhaite évaluer l’effet, ici le traitement) et au moins un effet dit “aléatoire” (la variable de regroupement, ici la boite). Les effets aléatoires ne sont pas évalués, ils servent seulement à indiquer au modèle que les données ne sont pas indépendantes pour une boite donnée. C’est ce qui permet à la déviance résiduelle d’être bien estimée, et ainsi à l’erreur standard des paramètres de ne pas être biaisée, et aux final d’obtenir des résultats fiables. En pratique, cela passe par l‘addition d’une quantité dans la matrice de variance- covariance des résidus.
Complexité
Les GLMM sont des approches plutôt complexes. Pour vous en convaincre, vous pouvez consulter cette FAQ; rédigée, entre autres, par Ben Bolker, un des grand spécialiste des GLMM.
Voici ce qu’il écrit en introduction :

Je ne vais donc pas ici rentrer dans les détails de cette approche, mais uniquement me concentrer sur les aspects pratiques.
Ce qu'il faut retenir
Je ne vais donc pas ici rentrer dans les détails de cette approche, mais uniquement me concentrer sur les aspects pratiques.
Trois éléments me semblent importants :
- C’est que le GLMM va nous permette de comparer les niveaux moyens des pourcentages observés pour les traitements A et B.
- La fonction de lien utilisée est la fonction logit, définie par :
\[ logit(p) = ln(\frac{p}{q}) = \sum_{j=1}^{p} \beta_j\;X_{ij} \]
Pour plus d’informations sur les fonctions de lien, je vous renvoie vers le tutoriel sur les GLM avec données de comptage.
- Ceci implique que pour estimer les proportions de cellules vivantes et mortes à partir des paramètres estimés par le modèle (le GLMM), il sera nécessaire d’employer la fonction inverse :
\[ p = \frac{ \exp^{(\sum_{j=1}^{p} \beta_j\;X_{ij})}}{1+exp^{(\sum_{j=1}^{p} \beta_j\;X_{ij})}} \]
Les données
Importation et structure
library(tidyverse)
library(here)
library(knitr)
mydata <- read.csv2(here::here("data", "Cellules.csv"))
head(mydata)
## vivant mort boite trt
## 1 138 24 B1 TrtA
## 2 94 14 B1 TrtA
## 3 46 8 B1 TrtA
## 4 38 3 B1 TrtA
## 5 78 3 B1 TrtA
## 6 91 2 B1 TrtA Le jeu de données est au format tidy avec :
- une colonne « vivant » : qui contient le nombre de cellules vivantes observées sur une image
- une colonne « mort » : qui contient le nombre de cellules mortes observées sur une image
- une colonne « boite » qui renseigne sur la boite dans laquelle l’observation à eu lieu
- une colonne « trt » qui renseigne sur le traitement reçu par les cellules.
Création d'une variable de proportion (freq_viv)
Les données ne contiennent pas de variable correspondant à la proportion de cellules vivantes. Cette variable sera utile pour représenter visuellement les données, nous allons donc la créer à l’aide de la fonction `mutate()` du package `dplyr` qui appartient au super package `tidyverse`.
mydata <- mydata %>%
mutate(freq_viv =(vivant/(vivant+mort)))
head(mydata)
## vivant mort boite trt freq_viv
## 1 138 24 B1 TrtA 0.8518519
## 2 94 14 B1 TrtA 0.8703704
## 3 46 8 B1 TrtA 0.8518519
## 4 38 3 B1 TrtA 0.9268293
## 5 78 3 B1 TrtA 0.9629630
## 6 91 2 B1 TrtA 0.9784946 On peut alors arrondir ces proportions, en utilisant la fonction round() comme ceci :
mydata$freq_viv <- round(mydata$freq_viv,3)
head(mydata)
## vivant mort boite trt freq_viv
## 1 138 24 B1 TrtA 0.852
## 2 94 14 B1 TrtA 0.870
## 3 46 8 B1 TrtA 0.852
## 4 38 3 B1 TrtA 0.927
## 5 78 3 B1 TrtA 0.963
## 6 91 2 B1 TrtA 0.978 Exploration des données
Pour avoir une première idée des données, le plus parlant est de réaliser une représentation graphique. Je choisis d’utiliser des boxplots en superposant les données. Pour cela, j’utilise le package `ggplot2` et ses couches `geom_jitter()` et `geom_boxplot()`. L’argument `width=0.25` permet aux points d’être contenus dans les boites. L’argument `alpha=0.5` permet d’avoir une couleur dans les boites plus claire que celle des points, et l’argument `outlier.alpha=0` permet aux outliers de ne pas être représentés deux fois (une fois par la couche geom_jitter et une fois par la couche geom_boxplot).
ggplot(mydata,aes(x=boite,y=freq_viv, fill=trt, colour=trt) )+
geom_jitter(width=0.25) +
geom_boxplot(outlier.alpha = 0, alpha=0.5) 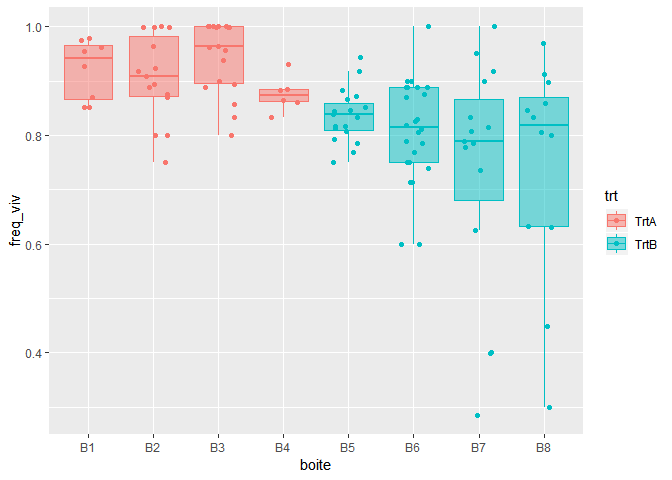
A première vue, le pourcentage de cellules vivantes est plus important en présence du traitement A qu’en présence du traitement B. L’analyse statistique, et utilisation du GLMM, nous permettra d’évaluer si cette différence est significative ou non.
On peut également résumer les données sous la forme d‘une table, en indiquant le nombre d’images prises par boite et le pourcentage moyen de cellules vivantes :
mydata %>%
group_by(trt, boite) %>%
summarise(nb=n(),
avg=mean(freq_viv))
## # A tibble: 8 x 4
## # Groups: trt [2]
## trt boite nb avg
##
## 1 TrtA B1 8 0.922
## 2 TrtA B2 15 0.906
## 3 TrtA B3 18 0.944
## 4 TrtA B4 6 0.876
## 5 TrtB B5 19 0.836
## 6 TrtB B6 24 0.808
## 7 TrtB B7 15 0.735
## 8 TrtB B8 12 0.744 Ajustement du GLMM
Création d'une variable combinée
Pour ajuster le GLMM, il est nécessaire de créer une variable combinée (nombre de cellules vivantes, nombre de cellules mortes). C’est cette nouvelle variable qui sera considérée comme la réponse dans le modèle :
mydata$y <- cbind(mydata$vivant, mydata$mort)
head(mydata)
## vivant mort boite trt freq_viv y.1 y.2
## 1 138 24 B1 TrtA 0.852 138 24
## 2 94 14 B1 TrtA 0.870 94 14
## 3 46 8 B1 TrtA 0.852 46 8
## 4 38 3 B1 TrtA 0.927 38 3
## 5 78 3 B1 TrtA 0.963 78 3
## 6 91 2 B1 TrtA 0.978 91 2
head(mydata$y)
## [,1] [,2]
## [1,] 138 24
## [2,] 94 14
## [3,] 46 8
## [4,] 38 3
## [5,] 78 3
## [6,] 91 2 Ajustement du modèle
Pour cela, on utilise la fonction glmer() du package lme4, en spécifiant :
- le traitement (variable trt) comme effet fixe,
- la boite comme effet aléatoire, avec la syntaxe `(1|boite)`,
- la fonction de lien logit et la distribution binomial des erreurs avec `family=binomial`.
library(lme4)
glmer1 <- glmer(y~trt+(1|boite), family=binomial, data=mydata)
summary(glmer1)
## Generalized linear mixed model fit by maximum likelihood (Laplace
## Approximation) [glmerMod]
## Family: binomial ( logit )
## Formula: y ~ trt + (1 | boite)
## Data: mydata
##
## AIC BIC logLik deviance df.resid
## 525.4 533.6 -259.7 519.4 114
##
## Scaled residuals:
## Min 1Q Median 3Q Max
## -4.8299 -0.5462 0.1386 0.8382 2.3740
##
## Random effects:
## Groups Name Variance Std.Dev.
## boite (Intercept) 0.01527 0.1236
## Number of obs: 117, groups: boite, 8
##
## Fixed effects:
## Estimate Std. Error z value Pr(>|z|)
## (Intercept) 2.2376 0.1090 20.537 < 2e-16 ***
## trtTrtB -0.8035 0.1349 -5.957 2.58e-09 ***
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Correlation of Fixed Effects:
## (Intr)
## trtTrtB -0.808 La fonction summary() renvoit plusieurs éléments. Les plus importants sont:
- des éléments de qualité du modèle (AIC, BIC, deviance, etc..),
- des éléments concernant les effets aléatoires,
- des éléments concernant les effets fixes,,
- la première ligne concerne le traitement A,
- la seconde ligne le traitement B, mais exprimé relativement au traitement A,
- la première colonne correspond aux estimations des paramètres,
- la seconde aux estimations des erreurs standard des paramètres,
- la troisième correspond à la statistique du test d’égalité à 0 des paramètres,
- la quatrième correspond à la p value du test.
Remarques:
- Les éléments des effets fixes sont exprimés dans l’échelle logit. Pour les estimer dans l’échelle des proportions, il sera nécessaire d’utiliser la fonction inverse du logit, décrite précédemment.
- Le fait que le coefficient relatif au traitement B soit <0 signifie que le niveau moyen des proportions de cellules vivantes est plus faible en présence du traitement B, par rapport au
traitement A. Pour savoir si nous pouvons avoir confiance dans la valeur de la p value, nous devons d’abord évaluer s’il existe une surdispersion.
Evaluation de la surdispersion
Il y a surdispersion (overdispersion en anglais) lorsque la variance des réponses observée est supérieure à la variance théorique, ici définie par la loi binomiale. Dans cette situation, l’erreur standard des paramètres est sous-estimée. Ceci peut conduire à une p value excessivement faible, et donc aboutir à une conclusion erronée. Il est donc important d’évaluer la présence d’une éventuelle surdispersion.
De manière grossière, la surdispersion peut s’évaluer par le rapport de la déviance sur le nombre de degrés de liberté, qui figurent sur la sortie de la fonction `summary(glmer1)`
surdisp <- 519.4/114
surdisp
## [1] 4.55614 Une surdispersion est mise en évidence lorsque le ratio est supérieur à 1 (autour de 1.5 par exemple). Le ratio étant ici de l’ordre de 5, on conclue à la présence d’une surdispersion.
Une autre approche, sous la forme d’une fonction, est proposée par Ben Bolker, ici :
overdisp_fun <- function(model) {
rdf <- df.residual(model)
rp <- residuals(model,type="pearson")
Pearson.chisq <- sum(rp^2)
prat <- Pearson.chisq/rdf
pval <- pchisq(Pearson.chisq, df=rdf, lower.tail=FALSE)
c(chisq=Pearson.chisq,ratio=prat,rdf=rdf,p=pval)
}
overdisp_fun(glmer1)
## chisq ratio rdf p
## 1.941786e+02 1.703321e+00 1.140000e+02 4.216640e-06 La p-value du test est < 0.05, on conclut également à la présence d’une surdispersion.
En cas de surdispersion, il est nécessaire d’utiliser une autre distribution des erreurs, telle que la structure « quasibinomial ». Celle-ci va conduire à une augmentation des erreurs standard des paramètres du modèle.
Utilisation de la loi quasi binomiale
Il n’est malheureusement pas possible d’utiliser la distribution « quasiobinomial » au sein de la fonction glmer(), puisque cela génère un message erreur :
glmer2 <- glmer(y~trt+(1|rep), family=quasibinomial, data=mydata)
Error in lme4::glFormula(formula = y ~ trt + (1 | rep), data = mydata, :
"quasi" families cannot be used in glmer Dans cette situation, deux solutions sont envisageables. La première consiste à employer la fonction `glmmPQL()` du package `MASS`. Dans ce cas, l’effet aléatoire doit être défini avec cette syntaxe : `random=~1|boite`:
library(MASS)
glmmPQL1 <- glmmPQL(y~trt, random=~1|boite, family=quasibinomial, data=mydata)
summary(glmmPQL1)
## Linear mixed-effects model fit by maximum likelihood
## Data: mydata
## AIC BIC logLik
## NA NA NA
##
## Random effects:
## Formula: ~1 | boite
## (Intercept) Residual
## StdDev: 0.08275886 1.320594
##
## Variance function:
## Structure: fixed weights
## Formula: ~invwt
## Fixed effects: y ~ trt
## Value Std.Error DF t-value p-value
## (Intercept) 2.2338383 0.1243801 109 17.95977 0.0000
## trtTrtB -0.7982217 0.1467616 6 -5.43890 0.0016
## Correlation:
## (Intr)
## trtTrtB -0.847
##
## Standardized Within-Group Residuals:
## Min Q1 Med Q3 Max
## -3.6902089 -0.4189964 0.1369458 0.6488932 1.8356178
##
## Number of Observations: 117
## Number of Groups: 8 On peut, voir que les erreurs standard des paramètres ont augmenté (par rapport à celles du modèle glmer1).
Ben Bolker propose également une autre méthode qui consiste à ajuster la table des coefficients, c’est-à-dire en multipliant l’erreur type par la racine carrée du facteur de dispersion et en recalculant les valeurs Z et p en conséquence. Le code suivant est donné:
cc <- coef(summary(glmer1))
phi <- overdisp_fun(glmer1)["ratio"]
cc <- within(as.data.frame(cc),
{ `Std. Error` <- `Std. Error`*sqrt(phi)
`z value` <- Estimate/`Std. Error` `Pr(>|z|)` <- 2*pnorm(abs(`z value`), lower.tail=FALSE) }) printCoefmat(cc,digits=3) ## Estimate Std. Error z value Pr(>|z|)
## (Intercept) 2.238 0.142 15.74 <2e-16 ***
## trtTrtB -0.804 0.176 -4.56 5e-06 ***
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1 Les erreurs standard sont alors encore plus élevées qu’avec le modèle glmmPQL. A présent que la surdispersion a été prise en considération, on peut analyser les résultats (ici par exemple ceux du modèle glmmPQL1 )
Résultats
summary(glmmPQL1)
## Linear mixed-effects model fit by maximum likelihood
## Data: mydata
## AIC BIC logLik
## NA NA NA
##
## Random effects:
## Formula: ~1 | boite
## (Intercept) Residual
## StdDev: 0.08275886 1.320594
##
## Variance function:
## Structure: fixed weights
## Formula: ~invwt
## Fixed effects: y ~ trt
## Value Std.Error DF t-value p-value
## (Intercept) 2.2338383 0.1243801 109 17.95977 0.0000
## trtTrtB -0.7982217 0.1467616 6 -5.43890 0.0016
## Correlation:
## (Intr)
## trtTrtB -0.847
##
## Standardized Within-Group Residuals:
## Min Q1 Med Q3 Max
## -3.6902089 -0.4189964 0.1369458 0.6488932 1.8356178
##
## Number of Observations: 117
## Number of Groups: 8 Comme expliqué précédemment, le fait que le coefficient relatif au traitement B soit < 0 signifie que le niveau moyen des proportions de cellules vivantes est plus faible en présence du traitement B, par rapport au traitement A.
La p value relative au traitement B correspond à la comparaison des niveaux moyens des proportions de cellules vivantes dans les deux traitements. Sa valeur < 0.05 nous indique que le niveau moyen des proportions de cellules vivantes en présence de traitement B est significativement plus faible qu’en présence de traitement A
Estimation des pourcentages de cellules vivantes
Comme expliqué précédemment, pour calculer ces pourcentages, il est nécessaire d’utiliser la fonction inverse du logit :
p_trtA = exp(2.23)/(1+exp(2.23))
p_trtA
## [1] 0.9029114
p_trtB=exp(2.23-0.80)/(1+exp(2.23-0.80))
p_trtB
## [1] 0.8069013 Estimation des intervalles de confiance des pourcentages
L’estimation des intervalles de confiance est plus difficile, car selon la paramétrisation du modèle, l’erreur standard de la seconde ligne est en réalité une erreur standard de différence.
Un moyen de ne pas se tromper est d’utiliser une autre paramétrisation du modèle (en ajoutant -1 à la fin des effets fixes). Dans cette paramétrisation, la seconde ligne n’est plus exprimée relativement à la première ligne. L’erreur standard est donc bien celle correspondant au niveau moyen prédit de proportion de cellule vivante en présence de traitement B. On peut alors s’en servir pour construire l’intervalle de confiance à 95% de cette proportion :
glmmPQL2 <- glmmPQL(y~trt-1, random=~1|boite, family=quasibinomial, data=mydata)
summary(glmmPQL2)
## Linear mixed-effects model fit by maximum likelihood
## Data: mydata
## AIC BIC logLik
## NA NA NA
##
## Random effects:
## Formula: ~1 | boite
## (Intercept) Residual
## StdDev: 0.08275886 1.320594
##
## Variance function:
## Structure: fixed weights
## Formula: ~invwt
## Fixed effects: y ~ trt - 1
## Value Std.Error DF t-value p-value
## trtTrtA 2.233838 0.12438015 6 17.95977 0
## trtTrtB 1.435617 0.07790088 6 18.42876 0
## Correlation:
## trtTrA
## trtTrtB 0
##
## Standardized Within-Group Residuals:
## Min Q1 Med Q3 Max
## -3.6902089 -0.4189964 0.1369458 0.6488932 1.8356178
##
## Number of Observations: 117
## Number of Groups: 8
# quantile 97.5 selon distribution de student
T <-qt(0.975,6)
T
## [1] 2.446912
binfA = 2.23-2.45*0.12
bsupA = 2.23 +2.45*0.12
IC_infA = exp(binfA)/(1+exp(binfA))
IC_supA = exp(bsupA)/(1+exp(bsupA))
IC_infA
## [1] 0.873912
IC_supA
## [1] 0.9258073
binfB = 1.44-2.45*0.08
bsupB = 1.44 +2.45*0.08
IC_infB = exp(binfB)/(1+exp(binfB))
IC_supB = exp(bsupB)/(1+exp(bsupB))
IC_infB
## [1] 0.7762595
IC_supB
## [1] 0.8369899 Ainsi, les proportions de cellules vivantes pour les traitement A et B, avec leurs intervalles de confiance à 95%, sont respectivement :
- 0.90 [0.87 ; 0.93] et
- 0.81 [0.78 ; 0.84]
Visualisation des résultats
colA<- "#FF7F00"
colB <- "#1E90FF"
mycol=c(colA, colB)
c("#FF7F00", "#1E90FF", "#FFFFFF")
## [1] "#FF7F00" "#1E90FF" "#FFFFFF"
ggplot(mydata,aes(x=boite,y=freq_viv, fill=trt, colour=trt) )+
geom_jitter(width=0.25)+
geom_boxplot(outlier.alpha = 0, alpha=0.5)+
scale_y_continuous(limits=c(0,1.5))+
scale_colour_manual(values=mycol)+
scale_fill_manual(values=mycol)+
geom_segment(x=0.75, xend=4.25, y=1.1, yend=1.1,col=colA)+
# geom_segment(x=0.75, xend=0.75, y=1.05, yend=1.1,col=colA)+
# geom_segment(x=4.25, xend=4.25, y=1.05, yend=1.1,col=colA)+
geom_segment(x=4.75, xend=8.25, y=1.1, yend=1.1,col=colB)+
# geom_segment(x=4.75, xend=4.75, y=1.05, yend=1.1,col=colB)+
# geom_segment(x=8.25, xend=8.25, y=1.05, yend=1.1,col=colB)+
geom_segment(x=2.25, xend=6.25, y=1.25, yend=1.25,col="black")+
geom_segment(x=2.25, xend=2.25, y=1.20, yend=1.25,col="black")+
geom_segment(x=6.25, xend=6.25, y=1.20, yend=1.25,col="black")+
annotate("text", x = 4, y = 1.3, label = c("p < 0.001")) 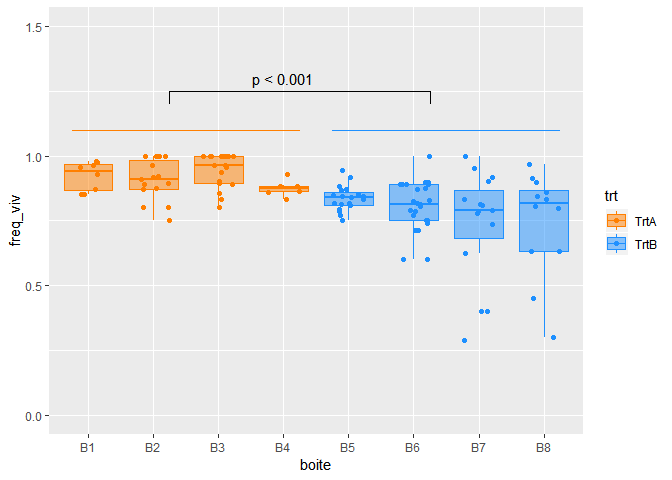
Pour aller plus loin
Si le sujet des GLMM vous intéresse, je vous recommande ces deux publications :
- Generalized linear mixed models: a practical guide for ecology and evolution (Ben Bolker et al.)
- A brief introduction to mixed effects modelling and multi-model inference in ecology (Harisson et al)
Voilà, j’espère que cet article permettra de vous familiariser avec les GLMM, et de comprendre dans quelles situations les employer.
Si cet article vous a plu, ou vous a été utile, et si vous le souhaitez, vous pouvez soutenir ce blog en faisant un don sur sa page Tipeee 🙏

Crédit photo : qimono
Crédit data : Rémi G 😉 (Merci !)

Très explicite 🤗
Merci Claire, très bon article.
Merci.un article très intéressant
Merci Claire , on vous adore.
Merci beaucoup pour cet article très intéressant.
Bonjour,
C’est un document important et par conséquence, je vous remercie vivement pour ce don gratuit de pleine utilité et de dévouement.
Avec mes encouragements pour Claire.
Fodil EL GAOUBI
Je n’arrive pas à exécuter les programmes de votre article. J’ai la version R-3.5.2-win
Bonjour,
Merci pour cet article
Lorsque je réalise cette analyse en l’adaptant à mes données j’ai le message d’erreur suivant
« fixed-effect model matrix is rank deficient so dropping columns / coefficients »
Je ne le comprend pas Pourriez vous m’expliquer ?
Bonjour,
un réponse ici : https://stackoverflow.com/questions/37090722/lme4lmer-reports-fixed-effect-model-matrix-is-rank-deficient-do-i-need-a-fi
Comme expliqué dans ce post, il est possible que certains niveaux d’une variable soient imbriqués dans certains niveaux d’une autre variable.
Bonne continuation
Bonjour et un grand merci pour votre travail et la qualité de vos articles.
Je souhaite bien comparer des proportions dans le cadre de mes recherches de thèse mais je ne suis pas certains que le GLMM soit bien adapté et je souhaiterai obtenir votre avis.
L’expérience se présente ainsi :
Je travaille sur des cultures de microalgues toxiques soumises à un traitement chimique.
Ces algues produisent 3 toxines différentes.
On prélève différents échantillons à différents temps après l’application du traitement.
J’ai donc:
– un facteur traitement
– un facteur temps sur des échantillons non indépendants car on prélève plusieurs fois dans la même culture au cours du temps.
– enfin chaque culture est réalisée en triplicat, ce qui n’est pas l’idéal.
Je souhaiterai déterminer si le traitement et potentiellement le temps on un effet sur le profil toxinique, autrement dit j’ai exprimé ma quantité de toxine par rapport à la somme des quantités des 3 toxines par cellules.
A un temps T, pour un traitement 1, j’ai donc des quantités de toxines exprimées comme-ci :
– Toxine A = 10% du total de toxine par cellule
– Toxine B = 80% du total de toxine par cellule
– Toxine C = 10% du total de toxine par cellule
Dans cette configuration est-ce bien le GLMM qui est adapté pour répondre à ma question ou suis-je à coté de la plaque ?
Merci encore.
Bonsoir,
à priori je partirai plutôt sur un modèle linéaire à effet mixte car votre variable réponse est un ratio de quantité qui est elle même une variable numérique continue non bornée. Les GLMM pour données de proportion c’est pour des ratio de données de comptages. Vous trouverez des exemples dans le R book (en cherchant un peu vous le trouverez en pdf 😉
Bonne continuation
Bonjour, Claire.
Merci pour cet article très intéressant et très clairement présenté.
Je voudrais savoir si la définition de l’odds ratio (OR) pour les Glm est valable et applicable pour les Glmm.
Par avance, merci.
Cordialement,
Hello Claire 🙂
J’avais exactement la même problématique que tu présentes au début de cet article, avec un facteur aléatoire de groupe, et un effet fixe dont je voulais calculer des intervalles de confiance, et ton article m’a énormément aidé !
Les modèles mixtes ça remonte à très loin, et je trouve que la plupart des articles sur les internets ressemblent à mes anciens cours de stats… pas très clairs 😀
Mais ton article est top et m’a beaucoup aidé !
J’ai encore quelques questions et je me demande si tu as des livres à recommander pour creuser la question (je vais déjà regarder les 2 articles que tu as proposés) et comprendre un peu mieux les maths et le R utilisés. Par exemple :
– Pourquoi ne peut-on pas donner les données brutes 0/1 en entrée ? Je remarque que ça donne des résultats proches mais pas identiques. Notamment au niveau de la détection de la surdispersion.
– Pourquoi et comment la surdispersion ? Je ne comprends pas d’où ça vient. Les données sont forcément issues d’une Bernoulli, et donc d’une binomiale quand on les ajoute, du coup.. pourquoi ?
Merci 😉
Hello Charles,
je suis ravie que l’article t’ait aidé.
Concernant les livres, ce que j’ai lu de plus compréhensible et de plus utile sur les LMM ou GLMM est dans le livre d’Alain Zuur : https://www.amazon.fr/Effects-Models-Extensions-Ecology-Author/dp/B007S7GBTM/ref=sr_1_1?__mk_fr_FR=ÅMÅŽÕÑ&keywords=alain zuur&qid=1578776225&sr=8-1
Tu verras aux avis sur Amazon, que je ne suis pas la seule à le trouver chouette !
En ce qui concerne l’utilisation des données brutes, ça veut dire que tu crées par exemple 138 lignes avec une réponse codée 1 et 24 lignes avec une réponse codée 0 (première ligne de mon exemple). Ensuite la fonction de lien est sensée être identique….je n’ai pas de réponse. Est ce que les petites différences sont sur les coeff ou sur les erreurs standard, ou les deux ?
Pour la surdispersion, ma compréhension est que le modèle calcule les erreurs standard des paramètres en faisant l’hypothèse que les données sont distribuées selon une distribution binomiale. Quand il y a surdispersion c’est que la dispersion réelle (cad observée) est supérieure à ce qui est attendue en théorie avec la distribution binomiale. Il faut donc corriger pour que l’erreur standard des paramètres soit calculée correctement, et donc en bout de chaîne que les tests soient exactes.
J’espère que ça t’aide 😉
Merci Claire pour cette belle explication, c’est très instructif
Bonjour, j’ai un script R qui me pose problème j’essaye de comprendre pour pouvoir le reproduire.
J’ai un souci en particulier avec cette ligne : (1 Variable_1 … Variable_n | species)
Pourrais-je avoir quelques explications . =)
Bonjour Claire,
j’apprécie bien les chapitres traités dans ton blog: félicitations!
Cependant, je souhaite apprendre à analyser dans R en utilisant lmer.
Pouvez-vous m’aider?
Merci d’avance
Bonjour,
Merci pour cet article !
J’ai cependant une petite question : qu’est-ce que le coefficient relatif < 0 cité dans la partie 7.Résultats ? Est-ce la value pour le trtB = – 0.7982217 ? ou la corrélation entre intercept et trtB = -0.847 ?
Merci 🙂
Bonjour,
Le coefficient relatif < 0 cité dans la partie 7 : Résultats, est trtB = – 0.7982217 . Bonne continuation
Bonjour !
Merci beaucoup pour ce cours, il est très bien expliqué.
Par ailleurs, il me semble que les résidus doivent se comporter normalement avec un GLM comme un GLMM, je voulais savoir comment procéder quand ce n’est pas vraiment le cas (transformation des données ? autre approche à utiliser ?).
Dans un de mes essais, je fonctionne par bloc de répétitions de 2 traitements (comme ton exemple) mais j’effectue mon comptage (vivant/mort) sur l’ensemble du bloc (et pas sur plusieurs zones comme toi avec tes photos). Je me demandais si cela changeais quelque chose au modèle ou si cela restait un GLMM ?
Merci d’avance pour ta réponse 🙂
Bonjour,
d’après ma compréhension, les résidus d’un glm ne doivent pas suivre une distribution normale puisque c’est un autre distribution des erreurs qui est employée (poisson, binomial, etc..).
Vous pouvez aussi allez voir du coté de package DHARMa développé par Florian Hartig : https://cran.r-project.org/web/packages/DHARMa/vignettes/DHARMa.html
Bonne continuation.
Bonjour Claire,
merci beaucoup pour cet article très bien écrit et très utile.
Je voulais vous demander si vous connaissiez par hasard une toolbox permettant de faire du Generalized NonLinear Mixed Model (GNLMM) ? J’ai une variable y binaire (0 1) et des prédicteurs organisé selon un modèle non linéaire.
Je sais que la librairie BRMS dans R est capable de le faire mais c’est de l’estimation Bayesienne (je n’ai rien contre le bayesien, mais je comprends mieux pour l’instant l’estimation par Maximum de Vraisemblance classique).
Merci d’avance !
Vincent
Bonjour,
ça semble possible avec la fonction gnlmm du package repeated, mais je ne les ai jamais utilisé.
Bonne continuation.
Bonjour Claire,
Merci beaucoup pour cet article très clair qui m’aide beaucoup pour la réalisation et la compréhension de mes statistiques de mon travail de fin d’étude.
J’ai réalisé la même démarche et avec la glmer, j’obtiens une surdispersion de 2,84 ce qui est de trop.. Je suis donc passé en glmmPQL :
glmmPQL(y ~Skyglow Position Skyglow*Position, random= list(Site=~1, Individus=~1, Jour=~1), family=quasibinomial(), data=mydata_freq)
où y correspond à mon nombre de feuilles endommagées corrigé par mon nombre de feuilles total (mydata_freq$y <- cbind(mydata_freq$Nombre_de_feuilles, mydata_freq$Nombre_de_feuilles_totales_endommagees))
mais je reçois ce message :
itération 1
Error in pdFactor.pdLogChol(X[[i]], …) :
NA/NaN/Inf in foreign function call (arg 3)
Je comprends bien que c’est parce que le calcul de ma fréquence n’est pas possible pour toutes mes observations et affiche Nan dans ma colonne « freq » et que cela se traduit par un zéro dans ma colonne y.
Je voulais donc savoir si il y avait une autre fonction qui prend en charge ce problème ? Ou faut-il le régler autrement ? Et si oui, avez-vous une idée ?
Merci d’avance !
Bonjour,
Là comme ça, non je ne sais pas trop quoi vous conseiller….
Je ne peux que vous renvoyer vers la page de Ben Bolker : https://bbolker.github.io/mixedmodels-misc/glmmFAQ.html
Bonne continuation
Merci pour l’article, mais peut t’on faire des comparaison multiples (fonction glht) sur les résultats du glmmPQL ?
Merci de votre aide.
Bonjour,
on dirait que oui :
library(MASS)library(nlme) # will be loaded automatically if omitted
str(bacteria)
fit <- glmmPQL(y ~ trt , random = ~ 1 | ID, family = binomial, data = bacteria)
library(emmeans)emmeans(fit, spec=pairwise~trt )
$emmeans
trt emmean SE df lower.CL upper.CL
placebo 2.19 0.386 49 1.418 2.97
drug 1.07 0.407 47 0.253 1.89
drug+ 1.49 0.415 47 0.655 2.32
Degrees-of-freedom method: containment
Results are given on the logit (not the response) scale.
Confidence level used: 0.95
$contrasts
contrast estimate SE df t.ratio p.value
placebo - drug 1.123 0.561 47 2.002 0.1229
placebo - (drug+) 0.705 0.567 47 1.245 0.4333
drug - (drug+) -0.418 0.581 47 -0.719 0.7535
Degrees-of-freedom method: containmentResults are given on the log odds ratio (not the response) scale.
P value adjustment: tukey method for comparing a family of 3 estimates
Bonjour Claire,
Merci pour cette page très bien expliquée. J’ai une petite question qui concerne le cas où la variable explicative possède trois niveaux (ou plus).
Dans cet exemple, la variable explicative trt n’a que deux niveaux, on ne peut donc comparer que ces deux niveaux (il n’y a qu’une ligne d’output qui compare le traitement B au traitement A).
Imaginons que la variable trt ait trois niveaux (A, B, C). Il y aurait deux lignes dans l’output, une qui compare B à A, et une autre qui compare C à A. On pourrait faire des tests post-hoc pour comparer les différents niveaux deux à deux, mais comment testerions nous si la variable trt dans son ensemble est significative ?
Merci d’avance !
Bonjour Maxime,
désolée de répondre aussi tardivement.
Si la variable explicative a 3 modalités, oui vous pouvez réaliser des comparaisons multiples, notamment avec le package emmeans.
Vous pouvez faire toutes les comparaisons deux à deux :
library(emmeans)emmeans(glmer, specs = pairwise ~ Var)
Ou tous les groupes vs un contrôle
emmeans(glmer, specs = trt.vs.ctrl ~ Var)En espérant que cela vous aide.
Bonne continuation
Bonjour !
Tout d’abord, un grand merci pour cet article !
J’aurais voulu savoir la signification précise du signe %>% et comment on pouvait le traduire pour avoir un RmarxDown parce que quand j’essaie d’en générer un, il bloque sur ce signe.
Un grand merci !
Bonne soirée à vous.
Bonjour
Le signe %>% est appelé pipe. Il vient du package tidyverse et permet d’enchainer les instructions avec une syntaxe simple.
Il faut donc charger le package tidyverse pour s’en servir :
library(tidyverse).
Bonne continuation
Bonjour,
Excellent article comme d’habitude, je vous en remercie
Je travaille pour la première fois sur les modèles crossover, du coup j’utilise les modèles linéaires mixte
• ID du sujet
• la séquence des traitements (AB ou BA)
• le traitement : A ou B
• la période (P1 ou P2)
• la réponse variable quantivive .
Je dois faire la même chose, en gardant ID, la séquence, le traitement, la période mais la variable réponse change
1er cas la réponse : évaluation de la douleur, une échelle numérique de 0 à 10
2ème cas la réponse : 2 modalités (oui/non)
3ème cas la réponse :5 modalités (croute, hématome, rougeur, inflammation, chaleur)
Est-ce que on est dans les modèles GLMM pour les 3 cas ?
Si c’est oui,
est ce qu’une échelle numérique de 0 à 10 considérer comme un comptage ou variable quantitative continues
2ème cas : est ce que GLMM régression logistique ?
3ème cas : 5 modalités je n’ai jamais croisé ce cas, du coup aucune idée sur le modèle
Merci d’avance
Bonjour Hocine,
Désolée de répondre aussi tardivement.
Pour le cas 1, je pense qu’un LMMM peut se tenter (vérifier que la normalité n’est pas trop éloignée des données), sinon une mixed effect ordinal logistique regression (je n’en ai jamais fait)
Pour cas 2 : oui GLMM logistique
Pour le cas 3 : si vous pouvez considérer les modalités comme ordinale : encore une mixed effect ordinal logistique regression, sinon, je ne sais pas !
Vous trouverez un peu d’informations sur ce papier : https://asja.springeropen.com/track/pdf/10.1186/s42077-020-00085-8.pdf
Dans tous les cas, je pense qu’une recherche biblio s’impose.
Bonne continuation
Bonjour!
J’ai réalisé un GLMM avec plusieurs variables explicatives. Dans ce cas, comment transformer les estimations des résultats ?
Pa
Par exemple si j’ai une fréquence de présence d’une fleur qui dépend à la fois du type de culture et du type de fertilisant ? Comment calculer la fréquence prédite en fonction des paramètres estimés par mon GLMM pour ces deux résultats ?
Merci d’avance!
Apolline
Bonjour,
Merci pour ce blog, une pépite !
J’ai une question à propos du choix de la variable combinée Y : Pourquoi faire un cbind(vivant,mort) au lieu d’un cbind(vivant,vivant+mort) ?
Pourtant le ratio correspond bien à freq_viv = (vivant/(vivant+mort)
Merci d’avance
Bonjour Thomas,
Il me semble que lorsqu’on réalise un glm, on peut utiliser deux syntaxes:
1) créer un vecteur double de type succès, echecs (ici vivant , mort) avec la commande y<- cbind(vivant mort) et ensuite utiliser la commande glm(y~var_explicative, family = "binomial, data = mydata) 2) créer un vecteur de proportions y <- vivant/(vivant+mort) et ensuite ajouter le nombre total de données (viant + morts) dans l'argument weights de la fonction glm : glm (y~var, weight= nb_total,family = "binomial, data = mydata). Il faut que nb_tot soit dans une colonne de mydata.Je pense que c'est pareil avec la commande glmer(). J'espère que cela vous aide. Bonne continuation
Bonjour, Merci pour ce beau article! cependnat j’ai une question, peut on utiliser des modéles linéaires mixtes avec des données de comptage?
Merci pour votre réponse
Bonjour,
avec des données de comptages les hypothèses de normalité et d’homogénéité des résidus se seront sans doute pas satisfaites, car les données de comptage suivent une distribution de poisson, qui stipule notamment que la variance est proportionnelle à la moyenne ; elle n’est donc pas constante (pas homogène). Dan sces cas là il faut employer un GLMM avec des données de comptages.
Bonne continuation.
Merci pour la clarté de cet article.
Quelle famille utilisée pour un modèle dont la variable d’interêt est un ratio borné en 0 mais pas en 1. Par exemple survie d’une cellule de type A/ survie d’une cellule de type B) sachant que ces survies sont exprimé comme des fréquences. Parfois la survie de A est meilleure (ratio >1) et parfois celle de B est meilleure (0<ratio<1).
Merci d'avance pour votre réponse, bonne continuation.
Bonjour,
je commencerais par essayer une loi gaussienne, donc réaliser un LMM avec la fonction lmer(), puis je vérifierais si les résidus suivent une loi normale.
J’espère que cela vous aide un peu…
Bonne continuation
Bonjour,
Merci pour cet article très pertinent.
Lorsque j’effectue le script, je tombe sur ça:
glmmPQL1 <- glmmPQL(y~Dominant.females+Non.dominant.females, (~1|Part.consumed), family=quasibinomial, data=mydata)
itération 1
Error in pdFactor.pdLogChol(X[[i]], …) :
NA/NaN/Inf in foreign function call (arg 3)
je voulais donc savoir à quoi cela est du ?
Bonjour,
je ne sais pas dut tout…
Bonne continuation
Bonjour Claire,
Un grand merci pour ces informations. A la lecture de votre article, je pense que cette procédure s’applique sur les données que je souhaite traiter, mais j’aurais voulu avoir votre avis à ce propos.
Mon jeu de données est le suivant :
40 individus répartis en 2 groupes (lot témoin et lot expérimental).
J’ai 8 mesures répétées sur ces individus (1 par semaine).
Les variables étudiées sont les suivantes : proportion d’observations où l’individu a été observé couché (ou proportion où l’individu a été observé en train de manger –> nb couche/nb obs sur la journee, avec le nb obs qui peut être légèrement différent à cause de données manquantes).
Je pensais soit faire comme vous indiquer dans l’article :
glmer1 et corriger pour la surdispersion
ou alors faire un modèle en utilisant la loi de Poisson, mias avec un offset pour prendre en compte le nombre réel d’observations.
Qu’en pensez-vous ?
Par ailleurs, il n ‘y a pas d’autres hypothèses à vérifier pour ce type d’analyse, si on est passé en quasibinomiale ?
Et pour les résultats, afin d’étudier les interactions qui m’intéressent, j’ai trouvé le code dans les commentaires et j’ai fait : emmeans(glmmPQL1, spec=pairwise~Lot|Semaine ) (afin de ne pas comparer mon traitement en semaine 1 avec mon contrôle en semaine 6, ce qui n’a aucun sens). Est-ce que ça vous parait correct ?
Merci d’avance pour votre retour.
Cordialement,
Marianne
Bonjour,
Dans mes données j’ai des proportions qui sont fait à partir de deux variables nombre de filles et nombre d’enfant total.
J’ai fait data$y<- cbind(data$fille , data$nb_enfant_total) est ce que c'est correct ? ou bien est ce que j'aurais du calculer une variable nombre de garçon et faire:
data$y<- cbind(data$fille , data$garçon) et ensuite mon modèle à partir de cet y là ?
Merci
Bonjour,
il faut faire data$y<- cbind(data$fille , data$garçon) et ensuite votre modèle à partir de cet y. Bonne continuation